

Tags
Anticipated results预期结果
SHAPE-MaP 允许在单核苷酸分辨率下测量 RNA 中大多数核苷酸的 RNA 灵活性。该实验产生高度可重复的定量信息,能够准确构建和检查 RNA-配体、RNA-RNA 和 RNA-蛋白质相互作用。成功完成该实验方案可产生深度测序文库(图5),该文库使用ShapeMapper的自动测序后处理进行解释,以产生最终的SHAPE反应性(图7和10)。这些 SHAPE 反应性为使用 SuperFold 建模 RNA 二级结构提供了一个起点,SuperFold 针对长序列(2,000 nt 或更长)进行了优化,但也适用于较短的 RNA 建模(图 11 和 12)。独特的实验工作流程经过优化,可探测小量、低丰度或复杂的 RNA 混合物。在这里,我们简要介绍了使用三种工作流程中的两种获得的代表性结果,即小RNA和随机工作流程。


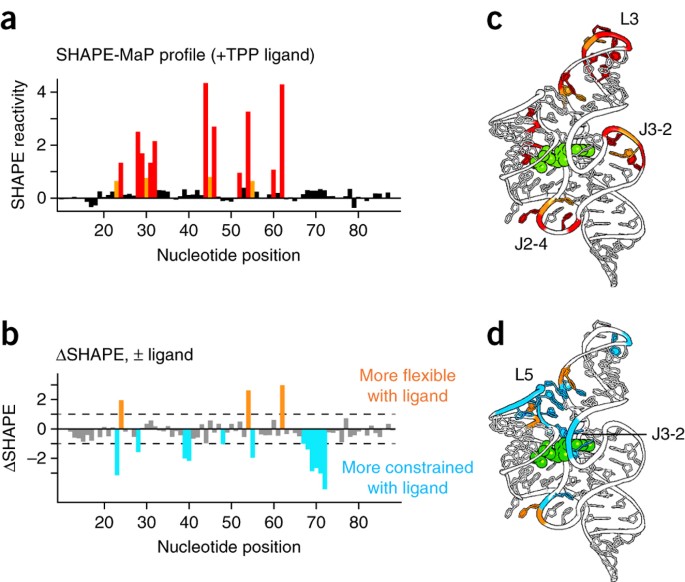

使用小 RNA 工作流程可以轻松获得 TPP 核糖开关适配体结构域的 SHAPE 图谱。使用这些数据,该核糖开关 RNA 的二级结构建模从仅使用最近邻热力学算法获得的碱基对预测精度 73% 提高到使用 SHAPE 定向建模的 96%28。观察到的反应性与基于配体结合RNA的局部核苷酸柔韧性的预期反应性非常一致(图10a,c)。反应性核苷酸位于构象灵活的单链区域,尤其是 L3 环和 J2-4 和 J3-2 链。总体而言,相对较少的核苷酸通过 SHAPE 产生反应,这与该 RNA 的高度受限构象一致。SHAPE-MaP还揭示了与TPP配体结合时引起的变化相对应的细微差异(图10b,d)。配体相互作用在 L5 环和配体结合口袋中的 J3-2 元件中诱导了一个大的结构组织。
大 RNA,例如细菌小核糖体亚基和大核糖体亚基 RNA(分别为 16S 和 23S),可通过将 Randomer 工作流程应用于总大肠杆菌 RNA 来轻松检查(图 13)。通过使用随机引物,两种RNA可以同时进行研究,并进行全自动分析,涉及∼3天的手动实验工作。主要的后处理要求是每个核苷酸的命中水平足够高,以便能够完全恢复底层 SHAPE 数据。一般来说,命中级别应为 5 或更大,对应于 1-2,000 的读取深度(参考文献 28)。
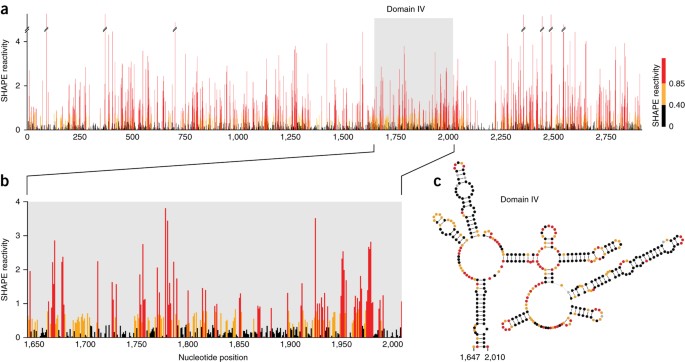

在计算数据处理后,仅 23S rRNA 亚基就代表了 ∼2,900 nt 的 SHAPE 反应性信息(图 13a)。将 23S rRNA 结构域 IV 的 SHAPE 反应性与公认的序列协变异衍生结构模型(图 13b,c)进行比较,显示出良好的一致性。参与规范碱基对的区域具有较低的 SHAPE 反应性,表明它们在结构上受到约束。相反,单链环和凸起区域具有高 SHAPE 反应性,表明结构灵活性。由于 MaP 方法固有的可扩展性,这些数据(跨越数千个核苷酸)在单核苷酸分辨率下与来自短 RNA(如 TPP 核糖开关)的数据一样准确。
ShapeMapper 分析完成后,呈现的 SHAPE 反应性配置文件提供了实验总体成功的清晰视图(.pdf“reactivity_profiles”文件夹中的文件)。一个成功的实验应该只有少量的负灰色条(表示没有数据点)和小的误差条(图7a)。
大多数SHAPE-MaP实验都取得了惊人的成功。获得的数据是可重复的、稳健的,与以前经过充分验证的 SHAPE 读数28 方法密切相关,能够进行一致的高精度 RNA 结构建模21 并恢复有关结构集成的详细信息48。在我们的实验室中,通常由不同的个体相隔数月进行完整的生物学重复,以产生相同的实验结果,误差范围很窄28。但是,当确实出现问题时,SHAPE 反应性配置文件以及其他 ShapeMapper 输出为故障排除和确定潜在原因提供了重要线索。
SHAPE-MaP实验失败的最常见原因是测序深度不足。测序到低深度的 SHAPE-MaP cDNA 文库不能产生高置信度的 SHAPE 反应性谱(如大误差线所示,图 7b),因此,它们无法进行准确的结构建模(参见原始 SHAPE-MaP 出版物28 中的图 3)。我们建议对每个实验条件的深度进行测序,读取深度高于 ∼2,000,以获得高置信度的 SHAPE 反应性和准确的结构模型。
通过检查 ShapeMapper 生成的配置文件,可以直接评估读取深度。对于使用小RNA工作流程的实验,需要相对平坦的深度分布,并且通常会实现(图8a)。带有凸起或阶梯的深度分布表明可能脱靶或非特异性引物结合。酶促片段化和标记 (Nextera) 导致转录本末端附近的读取深度较低,因为酶无法在双链 DNA 末端附近切割。此外,以这种方式制备的文库的读取深度曲线通常表现出尖锐性,这可能是由于碎片化步骤中的序列偏好造成的。扩增子工作流程导致转录本中间的轮廓相对平坦(图 8b),而随机过滤器工作流程通常产生不太均匀的深度轮廓(图 8c),因为引物结合的效率不同。低读取深度的区域,即使嵌入到中值读取深度高的区域,也无法通过SHAPE或任何其他结构探测方法产生可靠的反应性曲线。专门设计的引物可以改善或平滑具有AU富集区域的RNA的读取深度分布(图6)28。
ShapeMapper 生成多个直方图,这些直方图可用于区分成功和有问题的实验。例如,一个成功的实验将表明绝大多数核苷酸的读取深度都在2,000以上(图9a,中心),而一个不成功的实验(图9b,中心)可能具有较低的整体测序深度。成功的 SHAPE-MaP 实验还需要在背景之上进行足够水平的 SHAPE 修饰、有效的逆转录和无 DNA 污染。在成功的实验中(图9a,右),反应性大多为正,标准误差小于大多数反应性。成功实验的特征是无试剂样品的突变率在 0 到 0.2% 之间,加试剂样品的突变率强烈转向更高,而变性对照样品的突变率略有变化(图 9a,左)。图9d(左侧)显示了背景之上的低突变示例,可能是由于低水平的SHAPE修饰所致。嘈杂的SHAPE反应性曲线(例如,图7b)也表明实验失败。
SuperFold 分析流程为分析大 RNA 中的另一个挑战提供了解决方案:二级结构的自动可视化(图 12)。SuperFold 中实现了两种独立的可视化方法:(i) 圆图,它提供了公正的二级结构无模型视图,并能够快速可视化伪结,以及 (ii) 通过 Pseudoviewer59 网络服务提供的传统二级结构图。提供了连通性 (.ct) 和 XRNA (http://rna.ucsc.edu/rnacenter/xrna/) 文件,可用作其他结构可视化程序的输入。
融合的香农熵和SHAPE分析(图11)对于识别具有明确折叠的RNA区域以及识别值得进行详细功能跟踪分析的RNA部分特别有用。图中显示了预期输出的三个不同示例:大肠杆菌硫胺素焦磷酸适配体结构域、整个 16S rRNA 和整个 HIV-1 RNA 基因组(图 11)。同时包含低香农熵和低 SHAPE 反应性的区域似乎极有可能含有功能元件28,31。在扩展低香农熵/SHAPE区域以包括重叠的最小自由能螺旋后,几乎整个TPP核糖开关和核糖体RNA都包含低香农熵或低熵区域(图11,紫色阴影),这与它们作为RNA的已知角色一致,其功能需要特定的明确定义的折叠。相比之下,HIV-1 RNA基因组具有低香农熵和低SHAPE反应性的区域,并且还有许多区域预计不会形成明确定义的结构化状态(图11c)。配对概率弧说明了一组相对简单的 TPP 核糖开关和 16S rRNA 的可能碱基对(图 11a,b,见绿色弧)。相比之下,HIV-1 RNA基因组既具有明确确定的结构基序,也具有形成单一稳定结构可能性较低的区域(图11c)。
总之,SHAPE-MaP 以及 ShapeMapper 和 SuperFold 管道可产生定量核苷酸分辨率的 RNA 结构信息,实现准确的二级结构建模,识别大 RNA 中确定明确的区域,促进发现新型功能 RNA 基序,在单个实验中实现序列多态性的反卷积,检测配体和蛋白质结合的各种效应,轻松分析低丰度 RNA,并从短 RNA 优雅地缩放到转录组范围的分析,包括细胞中的转录组分析。我们预计 SHAPE-MaP 将有助于更深入地理解 RNA 结构和功能之间的关系。